
- Home
- La scuola
- Area Formazione
- Collegio Italiano dei Senologi
- Area donne
- Contatti
– Collegio Italiano dei Senologi: si ricorda che entro il 29 febbraio i Membri Effettivi devono provvedere al saldo della quota annuale di ammissione.
– Dal 10 febbraio sono disponibili online i programmi definitivi dei corsi validi come parte teorica del “Master in Ecografia Senologica” che si terranno a Palermo e Roma. Per informazioni e modalità di partecipazione clicca qui
– Programma definitivo della sessione di settembre (26-27-28) di “Building Bridges: the fully digital meeting in clinical oncology”. Evento patrocinato dalla Scuola Italiana di Senologia
– Inaugurato a Varese il VI Master Universitario di II livello in Senologia congiuntamente organizzato dall’Università dell’Insubria e dalla Scuola Italiana di Senologia. Il Master si rivolge ai laureati in Medicina e Chirurgia e si propone di fornire una preparazione specifica nel campo della Senologia che consentirà di formare una figura professionale che si faccia carico della valutazione diagnostico-preventiva e del trattamento specialistico in Senologia. La prossima edizione sarà organizzata nell’ambito dell’anno accademico 2024-2025
Dopo decenni di sostanziale immobilismo alla fine degli anni 70 la ricerca cominciò finalmente a produrre dati che portarono ad una svolta cruciale nelle strategie per il controllo del tumore della mammella. Nascevano allora la chirurgia conservativa, la radioterapia, la caratterizzazione biologica, i trattamenti adiuvante di chemioterapia o ormonali, ecc.. Con il consolidarsi dei risultati crebbe però anche la necessità di disporre di medici aggiornati per fare in modo che le nuove conoscenze e opportunità di cura non rimanessero confinate all’interno degli Istituti di ricerca, ma messe a disposizione del maggior numero possibile di pazienti in tutto il territorio nazionale.
 Da qui l’idea di Umberto Veronesi di dar vita nel 1984 alla nostra Scuola che, da allora, propone programmi formativi specifici dedicati a tutte le figure professionali impegnate quotidianamente nel contrasto della malattia. Abbiamo realizzato ad oggi oltre 1.700 eventi ed aggiornato e formato decine di migliaia di medici, tecnici di radiologia, infermieri, psicologi.
Da qui l’idea di Umberto Veronesi di dar vita nel 1984 alla nostra Scuola che, da allora, propone programmi formativi specifici dedicati a tutte le figure professionali impegnate quotidianamente nel contrasto della malattia. Abbiamo realizzato ad oggi oltre 1.700 eventi ed aggiornato e formato decine di migliaia di medici, tecnici di radiologia, infermieri, psicologi.
Qui è possibile trovare tutte le informazioni utili per poter partecipare agli eventi in programmazione (calendario, programmi, docenti e modalità d’iscrizione), consultare una rassegna della letteratura senologica internazionale aggiornata trimestralmente e avere un panorama delle novità editoriali in ambito senologico.
Calendario eventi in programmazione 2022
Master e percorsi di alta formazione
AIS – Attualità in Senologia: rassegna della letteratura
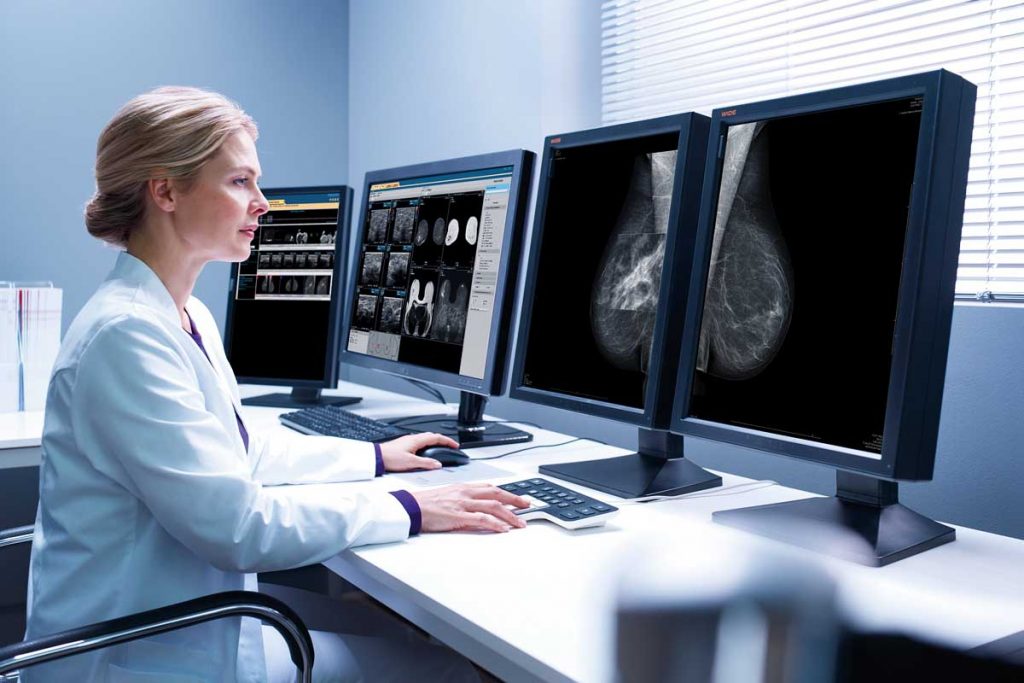
La Scuola, da oltre trent’anni impegnata a favorire la crescita professionale degli specialisti in radiologia che si dedicano alla senologia, propone una prova di refertazione mammografica, sviluppata e validata dal Consiglio Scientifico, basata su un campione significativo di esami eseguiti in donne asintomatiche sia nella pratica clinica quotidiana che in ambito di screening.
Il test fornisce un risultato in termini di autovalutazione mediante la stima della sensibilità e delle specificità a partire da una valore soglia determinato mediante validazione dello stesso test da parte di un campione di esperti (gold standard). Il raggiungimento di un punteggio soglia consente il rilascio di una certificazione di accuratezza diagnostica in mammografia. Leggi di più
“Ho voluto il Collegio perché credo che per un medico certificare la propria competenza in una disciplina così complessa come la Senologia sia un dovere verso le donne”. Umberto Veronesi
Il Collegio nasce nel 2012 per riunirvi i medici che dedicandosi alla cura della patologia mammaria dimostrano di avere e mantenere un elevato livello di aggiornamento professionale che permette di applicare con tempestività nella loro pratica clinica quotidiana le innovazioni della ricerca e garantire così alle pazienti i migliori trattamenti disponibili. Leggi di più
Regolamento e richieste di ammissione
Elenco Membri Certificati
I documenti del Collegio per la miglior pratica clinica
Nel corso degli anni molte delle nostre attività sono state rese possibili grazie alla collaborazione con alcuni dei più importanti Istituti Scientifici e Universitari italiani. Così come numerose Amministrazioni Pubbliche, Assessorati Regionali alla Salute ed Associazioni di Pazienti sono stati nostri partner in progetti di divulgazione scientifica volti a far crescere la cultura della prevenzione oncologica nella popolazione in generale e in particolare nelle persone più fragili (donne anziane, disabili, immigrate, detenute). Solo per citare due delle più recenti collaborazioni ricordiamo qui quella con All.Can International, organizzazione senza scopo di lucro multi-stakeholder che lavora per migliorare l’efficienza dei piani e delle politiche nazionali in ambito oncologico e quella con EUBREAST Study Group.
Sondaggio sulla formazione dei Senologi in Europa? European Union of Medical Specialists (UEMS-Multidisciplinary Joint Committee Breast Care) e European Breast Cancer Research Association of Surgical Trialists (the EUBREAST Network) promuovono un sondaggio aperto agli specialisti che si dedicano allo studio e alla cura dei tumori della mammella (radiologi, radioterapisti, patologi, chirurghi senologi, ginecologi, e oncologi) con l’intento di realizzare un quadro internazionale che faccia emergere l’eterogeneità in questo ambito. La Scuola Italiana di Senologia aderisce a quest’iniziativa e invita i medici che l’hanno frequentata a rispondere alcune domande sulla loro formazione a questo link.

Promuovere la consapevolezza che il tumore al seno può essere vinto grazie a una alleanza virtuosa fra medici e donne é sempre stato un nostro obbiettivo prioritario. Per far crescere nella popolazione la cultura della prevenzione abbiamo organizzato centinaia fra conferenze, incontri e dibattiti pubblici in tutto il territorio nazionale, offerto consulenze online gratuite, promosso campagne per la diagnosi precoce con visite senologiche, mammografie ed ecografie, prodotto e distribuito opuscoli e altro materiale informativo. Quest’Area del sito è un ulteriore strumento che mettiamo a disposizione delle donne italiane per informarsi, capire, fugare dubbi e perplessità ed aggiornarsi sulle nuove possibilità di diagnosi e cura.
Hai domande sul tumore al seno: scrivici qui!
“Paziente Diplomata” conferenza per donne con e senza tumore al seno: tutti i video
Tumore al seno: conoscere per vincere
Impareggiabile Maestro e caro Amico,
sono trascorsi ormai alcuni anni da quando te ne sei andato lasciando in tutti noi, che abbiamo avuto la fortuna di starti vicino e lavorare al tuo fianco, un vuoto davvero incolmabile. Ci hai sempre spronato a credere nel valore della scienza, della ricerca e dello studio. Ci hai dimostrato quanto l’empatia sia importante nel rapporto con i pazienti (ricordo che un giorno mi dicesti: vedi il sorriso è una medicina straordinaria, non costa nulla ma agisce tanto).

Ci hai insegnato la solidarietà, il rispetto per gli altri e per gli animali e tante, tante altre cose ancora. Ci hai però anche costretti, per anni, ad alzarci all’alba nell’illusoria speranza di riuscire ad arrivare in reparto o in sala operatoria prima di te ma tu, immancabilmente, eri già lì e questa è davvero l’unica cosa che non so se riuscirò mai a perdonarti.
Ti saluto qui nella Home della Scuola che hai voluto, fondato, sostenuto e amato certo che il tuo ricordo sia sempre ben vivo nella mente e nel cuore, non solo delle migliaia di pazienti che hai curato e a cui hai restituito la speranza, ma anche dei moltissimi medici che, come me, con i tuoi insegnamenti hai contribuito a rendere migliori.
Claudio Andreoli



